Article Text

Abstract
The clinical manifestation of frontal-variant Alzheimer’s disease (fvAD) is not typical, and it is difficult yet necessary to differentiate fvAD from frontal-variant frontal temporal dementia (fvFTD). We describe a patient with early-onset Alzheimer’s disease (AD) who presented with an fvFTD-like syndrome and apolipoprotein E ɛ3/ ɛ4 genotype. A brain amyloid imaging procedure, 11C-Pittsburgh compound B positron emission tomography (PET), supported the final diagnosis of AD. Our present case highlights the clinical variability that characterises early-onset AD. A multimodal approach is crucial when assessing rare forms of dementia.
- geriatric psychiatry
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Atypical frontal-variant Alzheimer’s disease (fvAD), where the brunt of Alzheimer’s disease (AD) pathology is found mainly in the frontal lobes, can present with a dementia syndrome more consistent with frontal-variant frontal temporal dementia (fvFTD) than AD. FvAD, which can take on disinhibited or apathetic forms, is a rare condition associated with early-onset AD.1 We describe here a patient with early-onset AD who clinically presented with fvFTD-like syndrome.
Clinical report and methods
A 52-year-old, right-handed woman with 19 years of education was first seen in our geriatric psychiatry department because of memory difficulties combined with progressive and prominent personality changes over the previous 3 years. She had previously been employed as a technician. Her medical history included a partial thyroidectomy due to a bilateral thyroid tumour 1 year ago.
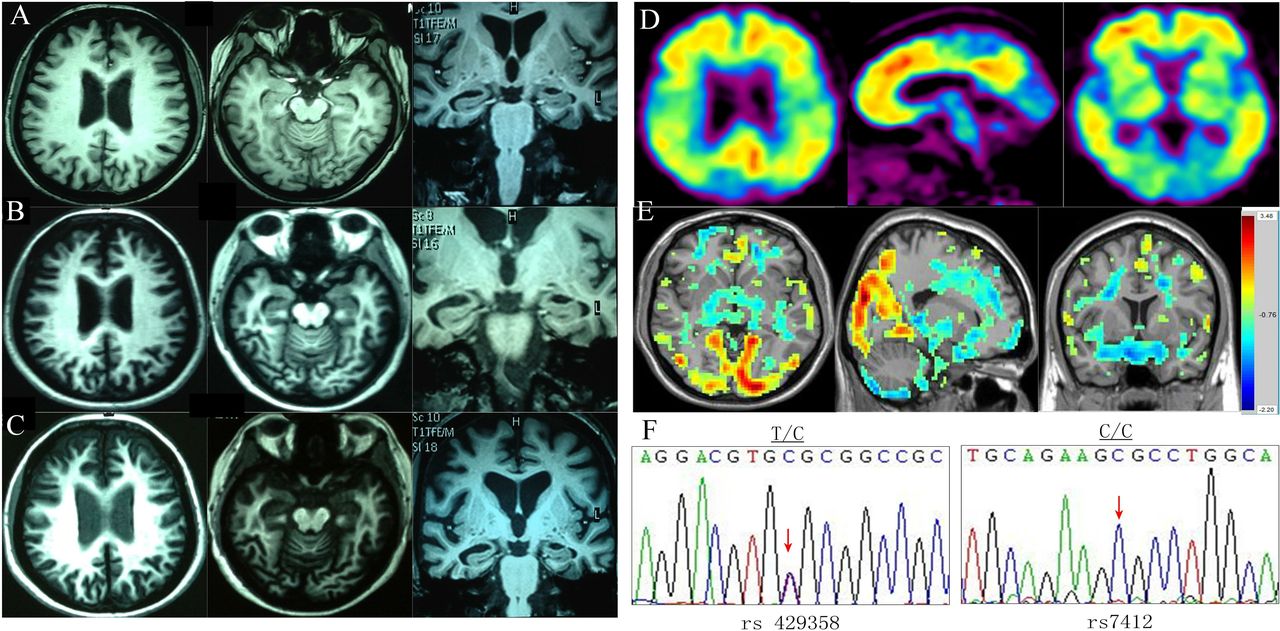
Her caregiver described her onset of memory impairment at age 49, which had developed insidiously. One year later, she showed apparent personality changes including irritability, anhedonia and apathy towards her family, accompanied with memory difficulties, calculation errors and speech disorganisation. She went to a local hospital for testing, and her first MRI scan in February of 2012 revealed mild global atrophy of the cerebral cortex and bilateral hippocampi, with frontal lobe predominance (figure 1A at age 50). The clinical examination revealed no apparent neurological deficits. Cognitive impairment with memory difficulties (Mini-Mental State Examination (MMSE)=22, Montreal Cognitive Assessment (MoCA)=20 at age 50) led to a suspicion of AD and, subsequently, therapy with Aricept was initiated. Six months later, the follow-up outpatient medical records showed that her abnormal behaviours had progressed. The patient became amotivated, lacking the initiative to visit her sick mother. She displayed unusual cooking habits, such as overseasoning of food. Around this time, combination therapy with memantine in addition to Aricept was started. One year later, her caregiver complained of aggravated symptoms, including sluggishness, slowed speech, delayed response, labile mood and odd behaviours such as random hiding of undergarments in bags and other places (MMSE=22, MoCA=18 at age 51). Her second MRI, performed in May of 2013, revealed increased global atrophy of the cerebral cortex and bilateral hippocampi when compared with the first MRI (figure 1B at age 51). At 3 years postonset of symptoms, the patient exhibited increasingly apathetic and strange behaviours, errors in simple arithmetic and poor memory to the extent that it affected her daily functioning (MMSE=12, MoCA=12 at age 52). This was reflected on the third MRI which revealed moderate global atrophy of the cerebral cortex with frontal lobe predominance and bilateral hippocampi shrinkage, leading to suspicion of FTD in March of 2014(figure 1C at age 52). One month later, she came to our department and received routine laboratory testing, which revealed no evidence of comorbid disease. Our Mini-Mental State Examination revealed a total score of 9 out of 30. The patient exhibited severe impairments in orientation (3/10), attention and calculation (1/5), recall (3/6), language (2/8) and visual construction (0/1). On the MoCA, the patient scored 8 out of 30. Deficits included visual construction (1/4), alternate line (0/1), recall (0/5), attention and calculation (2/6), language (0/6), abstraction (2/2) and orientation (3/6) (MMSE=9, MoCA=8 at age 52). The results of the remainder of her neurological examination were unremarkable. Brain MRI obtained in our department demonstrated moderate atrophy of the cerebral cortex and hippocampi, with frontal lobe predominance, similar to the third MRI done at the outside hospital. Genomic DNA was extracted from peripheral blood leucocytes and Apolipoprotein E (APOE) genotyping was performed at the locus of rs429358 and rs7412 which determined the three ɛ alleles that are established genetic risk factors for late-onset AD. The direct sequencing showed T/C heterozygous form at the locus of rs429358 and C/C homozygous form at rs7412, which identified APOE ɛ3/ ɛ4 genotype (figure 1F). In addition, we tested three pathogenic genes for early-onset AD including amyloid precursor protein (APP), Presenilin-1 (PS1) and Presenilin-2 (PS2) using the direct sequencing method, and we did not find any mutation in the three genes. The regional homogeneity (ReHo) analysis of blood oxygen level-dependent (BOLD) functional imaging data showed decreased ReHo (blue) in frontal lobes (figure 1E). Due to apparent personality changes and behaviour dysfunction in the early stages of the disease and late-onset AD risk genotype of APOE ɛ3/ ɛ4, the patient underwent 11C-Pittsburgh compound B (PIB) PET to provide clarification of diagnosis. This revealed elevated PIB binding in diffuse cortical areas, particularly in the bilateral frontal, parietal, occipital, temporal cortices and posterior cingulated gyrus (figure 1D). The 11C-PIB PET, combined with memory dysfunction as the first clinical manifestation and MRI, confirmed the final diagnosis of AD.
{kind=link}
Brain imaging and gene detection of the patient. (A) The patient’s MRI from the first year revealed a mild atrophy of the whole cerebral cortex and bilateral hippocampus dominated in the bilateral frontal lobes, basal ganglia and brainstem (time of repeatation(TR): 9.8, time of echo(TE): 5.8, at age 50). (B) The patient’s MRI from the second year revealed more atrophy of the whole cerebral cortex and bilateral hippocampus than the first MRI (TR: 9.8, TE: 4.6, at age 51). (C) The patient’s MRI from the third year revealed moderate atrophy of the whole cerebral cortex with frontal lobes more affected and bilateral hippocampus (TR: 9.8, TE: 4.6, at age 52). (D) 11C-PIB PET revealed increased PIB uptake in diffuse cortical areas (at age 52). (E) The ReHo analysis of BOLD functional imaging data showed decreased ReHo (blue) in the frontal lobes. (F) DNA sequence at the locus of rs429358 and rs7412 of APOE gene from the patient. The red arrow indicates T/C heterozygote and C/C homozygote of two single nucleotide polymorphisms (SNPs), respectively (rs429358 and rs7412). APOE, apolipoprotein E; BOLD, blood oxygen level-dependent; PIB, Pittsburgh compound B; ReHo, regional homogeneity.
Discussion
The patient exhibited insidious memory decline as the first clinical manifestation. The initial MRI revealed a mild global cerebral cortex and bilateral hippocampi atrophy with frontal lobe predominance, and APOE ɛ3/ ɛ4 genotype showed 3–4 times increase in suffering from late-onset AD.2 These findings are suggestive of a diagnosis of AD; despite this, we did not find the mutation of early-onset AD pathogenic genes including APP, PS1 and PS2. With disease progression, the patient experienced more prominent personality changes in the early stages of the disease. MRI at this stage revealed moderate atrophy of the cerebral cortex and bilateral hippocampi, with frontal lobe predominance. There was similar result in that ReHo value decreased in the frontal lobes through the analysis of BOLD functional imaging data. Despite combination therapy with a cholinesterase inhibitor (AChEI) and a non-competitive N-methyl-D-aspartate receptor antagonist (Memantine), cognitive function continued to decline rapidly over the course of 3 years. These particular symptoms seemed suggestive of a diagnosis of FTD. Although the patient began to show mild memory decline and errors in calculation in the early stages, she also exhibited prominent deterioration in personality and social etiquette in the first 2 years. Symptoms indicative of frontal lobe pathology included abulia, apathy, impaired verbal fluency and execution dysfunction. We also considered Lewy body dementia (LBD), given the patient’s abnormal emotional behaviours, but excluded this possibility due to the lack of a Parkinson-like syndrome and definitive hallucinations. Another possibility was a form of rapidly progressive dementia such as prion protein disease. However, the patient was without deficits upon neurological examination or characteristic abnormalities (‘cortical ribbon sign’) on MRI. Beta amyloid is a common biomarker of AD. PIB can specifically bind to extracellular and intravascular fibrous Aβ, allowing PIB PET to be used as an Aβ-specific molecular probe to assess the deposition of Aβ plaques in different brain regions. It can also be used as a non-invasive test method to help dynamic monitoring of protein deposition, playing an important role in early diagnosis of patients with AD. Finally, the patient’s 11C-PIB PET result confirmed the diagnosis of early-onset AD, given her memory decline as the first clinical manifestation, obvious cortex and hippocampus atrophy on MRI, diffuse cortical dysfunction supported by neuropsychological examinations during advanced stages of the disease and increased PIB binding in diffuse cortical areas.
FTD is a neurodegenerative disease that affects the frontal and/or temporal lobes, typically affecting individuals between the ages of 45 and 65 years.3 Frontal-variant FTD (fvFTD) is the most common form of FTD and is characterised by progressive and prominent deterioration in personality, which takes disinhibited or apathetic forms.1 3 Atypical frontal-variant AD (fvAD), in which the brunt of AD pathology including neuritic plaques and neurofibrillary tangles are found mainly in the frontal lobes, can present with a dementia syndrome more consistent with fvFTD than AD.1 Our patient appeared to match the apathy symptom profile, which seems to be a behavioural marker indicating a more severe and aggressive clinical decline, with faster cognitive, functional and emotional progression.4
The following signs may indicate a clinical diagnosis of fvAD in patients presenting with a fvFTD-like syndrome: genetic signs for AD, including positive family history and APOEε4 allele, and impaired episodic memory performance. As far as laboratory testing and imaging, it may be beneficial to test for cerebrospinal fluid biomarkers or use amyloid imaging procedures such as PIB PET, which aid in the differentiation between fvAD and fvFTD. Tun et al5 thought apathy syndrome in AD independently increased the risk of mortality. In typical AD, treatment with AChEI and/or memantine has been shown to positively affect cognitive, behavioural and daily functioning. In fvAD, among several psychoactive agents proposed to treat apathy, the best results seem to have been obtained with AChEI.6 Selective serotonin reuptake inhibitors (SSRI) are still a controversial therapeutic approach to treat fvAD. In the case of our patient, however, AChEI seemed ineffective in delaying disease progression.
Behavioural and psychological symptoms of dementia (BPSD) are major and common syndromes in demented persons. FTD was characterised by a much younger mean age of onset than AD and other types of dementia in previous observations. Patients with AD often manifest motor behaviour disturbance, aggressiveness, mood disturbances and psychotic symptoms. Activity disturbances were noted in almost all patients (92.3%) with FTD. Although researchers summarised the clinical characteristics of BPSD in patients with AD and FTD, there remains the possibility of misdiagnosis through observational data. Beach et al assessed the accuracy of the clinical diagnosis of AD based on autopsy results, or the ‘gold standard’, and demonstrated that depending on the permissiveness of clinical and neuropathological criteria, sensitivity ranged from 70.9% to 87.3% and specificity ranged from 44.3% to 70.8%.7 Some studies have found that between 12% and 23% of patients diagnosed with AD do not have sufficient AD pathology at autopsy to account for the presence of dementia (‘misdiagnosed’).8 Improving diagnostic accuracy in clinical settings may help reduce unnecessary treatment as well as increase the administration of appropriate therapy for patients’ conditions. As a very effective biomarker, beta amyloid PET scan can lead to substantial changes to the methodology of providing diagnoses and intended management plans.9 10
Conclusion
FvAD is a rare form of AD which can masquerade as an fvFTD-like syndrome. In the reported case, neither clinical examination nor MRI could definitively differentiate fvAD from fvFTD. Ultimately, these tools, in combination with PIB PET, allowed us to make the diagnosis of fvAD. A multimodal approach is therefore crucial when assessing rare forms of dementia.
References
Han Cai is an attending physician of geriatric psychiatry at Wuhu Fourth People’s Hospital in Anhui province, China. She obtained a master's degree in Mental Health from Anhui Medical University in China in 2013 where she built animal models in order to study immune changes associated with schizophrenia. Cai is currently chairing an in-hospital project researching the relationship between smoking, and metabolic diseases and cognitive function. She is now studying at the Department of Geriatric Psychiatry in Shanghai Mental Health Center in China. Her research interests include geriatric psychosis, especially Alzheimer's disease.

Footnotes
HC and SN are joint first authors.
HC and SN contributed equally.
Deceased 6
Contributors SN provided the case report part. HC finished the writing of the manuscript. WL performed the literature search and LS provided critical revision for the manuscript. SN, HC, WL, XL, LS and SX contributed to and approved the final manuscript.
Funding This study was supported by a grant of Clinical Research Center Project of Shanghai Mental Health Center (CRC2017ZD02), Natural Science Foundation of China (81301139) and Western medical guidance project of Shanghai science and Technology Commission (17411970100).
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.