Article Text

Abstract
Background Although recent data in healthy humans suggestthat treatment with intranasal oxytocin (OT) may facilitate extinction recall,to date, little is known about the effects of OT on memory consolidationprocesses.
Aim To examine the effect of intranasal administration of OT compared with placebo on memory consolidation blockade of a de novo fear memory in a classical 2-day fear conditioning procedure.
Results There were no significant differences between the OT and the placebo groups on the first two extinction trials (mean (SD)=0.01 (0.39) vs 0.15 (0.31), t=−1.092, p=0.28). Similarly, during early extinction, analysis of variance for repeated measures failed to show significant main effects of extinction trials: trials (F(4, 112)=1.58, p=0.18), drug (F(1, 112)=0.13, p=0.72) or drug × trials interaction (F(4, 112)=0.76, p=0.56).
Conclusion Our results suggest that OT administered in a double-blind fashion immediately after fear conditioning does not significantly reduce consolidation of fear learning as measured by a differential skin conductance response tested at the beginning of extinction.
- fear conditioning
- oxytocin
- extinction learning
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Post-traumatic stress disorder (PTSD) is a distressing and debilitating condition that may develop after exposure to a traumatic event.1 Although large epidemiological studies suggest that up to 25% of individuals exposed to a potentially traumatic event develop PTSD,2 3 there is a paucity of efficacious prevention strategies. Recent findings suggest that early psychosocial cognitive behavioural interventions initiated within a few hours after trauma exposure might be effective in decreasing the risk of developing PTSD.4 However, more research is needed to understand how to optimise early intervention in an effective fashion that can be broadly disseminated into acute care settings and does not require highly specialised trained therapists. Another proposed strategy for secondary prevention of PTSD is thus to use pharmacological agents in the acute aftermath of trauma to interrupt the overconsolidation of trauma memories. In addition to failures of extinction, memory overconsolidation has been suggested to be a key process underlying the pathophysiology of PTSD.5 To date, a few studies have attempted to block the putative consolidation of memories by administering a pharmacological agent within hours of a traumatic event, with mixed results. In humans, the beta-blocker propranolol has been the most studied compound for memory consolidation blockade. While results from two initial studies were promising,6 7 more recent studies have failed to show efficacy for early administration of propranolol in preventing PTSD development.8 9 More research is needed however to understand if alternative pharmacological interventions may be more effective, or if the strategy of interruption of memory consolidation with pharmacotherapy is not an effective strategy overall.
Oxytocin (OT) is a nine-amino-acid peptide that is synthesised in the paraventricular and supraoptic nuclei of the hypothalamus and projected to the posterior pituitary and limbic areas including the hippocampus, amygdala, striatum, hypothalamus, nucleus accumbens and locus coeruleus.10 While OT was first reported to be involved in labour, lactation and maternal care across species, more recent animal and human studies found that it was also implicated in social behaviours, anxiety (for review see ref 11), stress-related disorders,12 and memory formation and consolidation.13 For example, OT administration, compared with placebo, immediately after a learning trial in a passive avoidance task was associated with a decrease of the subsequent avoidance behaviour in rats, suggesting that OT may have weakened the memory of the experience.14 Further, Hou et al15 demonstrated that OT administered to rats immediately after reactivation of a conditioned fear memory also impaired reconsolidation of the fear memory.
In humans, early findings suggest that intranasal OT (24 IU) administered before conditioning may facilitate the acquisition of a conditioned fear response.16 However, intranasal administration of OT (24 IU) immediately after fear acquisition has been found to decrease affective evaluation of conditioned faces, compared with placebo,17 suggesting that OT might alter consolidation of the fear memory; however, memory recall took place 45 min after OT administration in this study. Because OT has been suggested to have an anxiolytic effect,18 OT may have influenced fear expression, rather than the fear memory. Similarly, Eckstein et al19 in a 1-day fear conditioning procedure found that intranasal OT administered immediately after fear acquisition (and before extinction) was associated with enhanced extinction learning. While these results suggest that OT may enhance extinction learning, it is also possible that OT weakened fear consolidation and thereby promoted faster extinction. To date, no studies have specifically examined the effect of intranasal OT administered immediately after fear acquisition on memory consolidation. Such a study would have to test fear recall after sufficient time has elapsed to rule out the possibility of an anxiolytic effect of OT on fear expression.
The present study examined the effect of intranasal administration of OT compared with placebo on memory consolidation blockade of a de novo fear memory in a classical 2-day fear conditioning procedure. OT is a well-tolerated generically available agent20 with the potential for clinical utilisation for secondary prevention of PTSD, should it be effective in blocking memory consolidation. We hypothesised that treatment with intranasal OT 30 IU immediately after fear conditioning would be associated with reduced consolidation of the conditioned fear memory as evidenced by a smaller differential skin conductance response (SCR) during the first two extinction trials, compared with placebo. SCR was chosen as outcome as it has been extensively used in prior research studying fear expression.21
Methods and materials
Participants
Participants were recruited through local advertisement and included healthy adults aged 18–65 years. Based on a prior finding that low conscientiousness was associated with greater likelihood of acquiring conditioned responses in the laboratory,22 participants were required to score less than 42 on the conscientiousness subscale of the NEO Five Factor Personality Inventory.23 Any current Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) Axis I diagnosis or attention deficit/hyperactivity disorder, as well as a lifetime diagnosis of psychotic disorder, bipolar disorder or obsessive compulsive disorder as measured by the Structured Clinical Interview for DSM-IV (SCID-IV24), was exclusionary. Women of childbearing potential not using medically accepted forms of contraception, with a serious medical condition including head injury and seizures, using any supplemental hormones (birth control, oestrogen, testosterone, prednisone and so on), psychotropic medication or narcotics, with a history of hyponatraemia, or with known hypersensitivity to OT or any of the excipients of Syntocinon Nasal Spray were also excluded.
Study drug
Participants were those who met the entry criteria, completed baseline questionnaires and underwent conditioning procedures. After completing the conditioning procedure, participants were administered intranasal OT 30 IU or placebo saline spray in 1:1 randomised, double-blind fashion. This dose is comparable with that used in other human studies of intranasal OT.25 26 The nasal sprays were delivered in a high-precision metered-dose spray vial, which participants self-administered with 10 puffs into the nostrils (approximately 3.08 IU OT per spray).
Blinding and randomisation
The randomisation list was generated by our research pharmacy. The pharmacist manually randomised subjects based on this list. All other study personnel, including investigators and research coordinators, as well as the participants, remained blinded throughout the study.
Psychometric measures
Participants were screened by trained clinical raters using the structured interview for DSM-IV (SCID-IV24) and the attention deficit disorder / attention deficit hyperactivity disorder module of the MINI Plus (Mini-International Neuropsychiatric Interview).27 On day 1, just prior to undergoing the experimental paradigm, participants were also assessed with self-rated measures including the Beck Anxiety Inventory,28 the State-Trait Anxiety Inventory29 and the Anxiety Sensitivity Index,30 as well as with the Penn State Worry Questionnaire31 and the Beck Depression Inventory.32 These allowed us to ensure baseline levels of anxiety (potential confounds) did not differ across groups.
Conditioning procedures
Because of its wide application to human aversive conditioning (eg, refs 33 34), SCR was the outcome measure of primary interest. The experimental procedures followed closely those of Bui et al,34 and were conducted in a sound-attenuated, temperature-controlled and humidity-controlled room. The conditioned stimulus (CS) were displayed on a monitor positioned 1.2m in front of the participant. The CS+ and CS− were represented by a yellow circle and a white square, respectively. The unconditioned stimulus (UCS) was a 500 ms electric shock delivered through the electrodes attached to the second and third fingers of the dominant hand and generated by a Coulbourn Transcutaneous Aversive Finger Stimulator (E13-22; designed for human conditioning research) set at a level determined by the participant to be ‘highly annoying but not painful’. Prior to the conditioning procedure, participants’ skin conductance (SC) was tested for fear conditioning viability. Viability was defined as an SC level greater than 0.3 μSiemens at baseline and a minimum increase of 0.23 μSiemens when performing mental arithmetic.
On day 1, the participant was seated in a comfortable armchair, and physiological recording electrodes and those for administering the shock UCS were attached. Prior to setting the UCS level, the research coordinator gave the following instructions: ‘Forthis experiment, you will set your own level of electric stimulation. You should choose a level that is highly annoying but not painful. I will start the stimulation at a very low level and gradually increase the level until you say stop. The level that you set will then be used throughout the remainder of the experiment’. The research coordinator then set the UCS level (ranging between 0.2 and 4.0 mA) and noted it. Once the UCS level was established, the participant was instructed to relax during a 5 min baseline period and received instructions about the upcoming phases of the study.
The experiment was then administered by the computer. First, there was a 5 min baseline period. Habituation consisted of five presentations each of the to-be CS+ and CS− in pseudo-random order, with no more than two consecutive presentations of the same stimulus type. The CS duration was 8 s, and the intertrial interval was 20±5 s, determined at random by the computer. The acquisition phase (ACQ) consisted of five CS+ and five CS− trial presentations with a 500 ms shock pulse (UCS) occurring immediately following each CS+ offset (100% reinforcement). SC level was sampled at 1000 Hz beginning 2 s prior to CS onset and ending 6 s after CS offset.
After the conditioning procedure was completed, intranasal OT 30 IU or saline placebo spray was administered in a 1:1 randomised, double-blind fashion. On the second day of the study, that is, the extinction phase, participants were assessed at the same time of day and using a similar procedure as for the first day, except that there was no shock-setting procedure, no drug administration and it took place in a different room (‘context’) from ACQ. The extinction phase consisted of 10 non-reinforced presentations each of the CS+ and CS− (only the first five were analysed for the present study). The first two extinction trials were used as a measure of fear acquisition and the primary outcome for the trial. Finally, at the end of the extinction phase, participants were asked to identify the particular stimulus (yellow circle or white square) paired with the shock.
Physiological equipment
We used a Coulbourn Lablinc V, Human Measurement System (Coulbourn Instruments, Allentown, Pennsylvania) to record SC level, which has been widely used as a primary outcome measure in prior conditioning work.33 Specifically, Coulbourn-isolated SC coupler (V71-23) using a constant 0.5 V through 9 mm (sensor diameter) was used to assess SC levels. In Vivo Metric Ag/AgCl electrodes were placed on the hypothenar surface of the participant’s non-dominant hand, 14 mm apart, as determined by the width of the adhesive collar.35 The SC analogue signal was digitised by a Coulbourn analogue to digital converter (V19-16). A notebook computer with custom-designed software was used to display the CS and to sample and store digitised physiological signals.
Data reduction
Failure to show a mean SC unconditioned response (UCR) greater than 0.1 μSiemens to the five presentations of the UCS on day 1 indicated SC non-responsiveness. These SC non-responders were excluded from all analyses, as they would be expected not to show a conditioned response. In addition, participants who failed to show a measurable conditioned response defined by a mean SC conditioned response to the five CS+ trials in the ACQ greater than or equal to 0.05 μSiemens were also excluded from all analyses. Finally, individuals who did not exhibit differential conditioning as evidenced by a differential SCR averaged across acquisition trials ≥0.1 μSiemens were also excluded.
An SCR score was calculated for the CS interval of each trial by subtracting the mean SC level during the 2 s immediately prior to CS onset from the highest skin conductance level (SCL) recorded during the 8 s CS duration. An SCR score for the interval containing the UCR was calculated by subtracting the average SC level during the final 2 s of the CS interval from the maximum SC level during the 6 s interval following CS offset. These scoring procedures followed those used by Orr et al.33 Finally, the differential SCR (primary outcome) was calculated as the SCR for the CS+ interval minus the SCR for the CS− interval for each pair of successive CS+ and CS− trials.
Statistical analysis
χ2 tests and Student’s t-tests for independent samples were used to compare sociodemographic variables (age, gender, race and ethnicity) and psychometric measures (anxiety, depression and traits) between the two groups (OT vs placebo). The differential SCR values were square root-transformed to achieve normality prior to analyses. The SC resting level was determined by calculating the mean SC level during the 5 min baseline rest period. An SC orienting response was calculated by averaging each participant’s SCR to the first presentation of the CS+ and CS− during habituation.
Student’s t-test and a multiple regression controlling for covariates were used to compare the primary outcome (differential SCR during the first two extinction trials) between individuals randomised to intranasal OT versus placebo. In addition, a series of analyses of variance for repeated measures (ANOVAR) were conducted to examine SCR across trials separately by phase (habituation, acquisition, extinction). All analyses were conducted with Stata V.12,36 and the level of statistical significance was set to 0.05 (two-tailed).
Results
Demographics and psychometrics
Of the 73 participants who met the eligibility criteria, 60 started the fear conditioning paradigm. Of those, 8 were excluded from the analyses for SC non-responsiveness (absence of an SCR to the UCS) and an additional 10 for failure to show an SCR to the CS+ during the ACQ. Twelve participants were also excluded because they failed to exhibit a differential SCR during the ACQ that met the ≥0.1 μSiemens criterion. Thirty participants met each of these SC criteria and were included in analyses presented in this manuscript. Of these 30 participants, 14 had been randomised to OT and 16 to placebo. The flow chart of participants is reported in figure 1. Sociodemographic variables (sex and age) and clinical variables (anxiety, depression and traits) are presented in table 1.
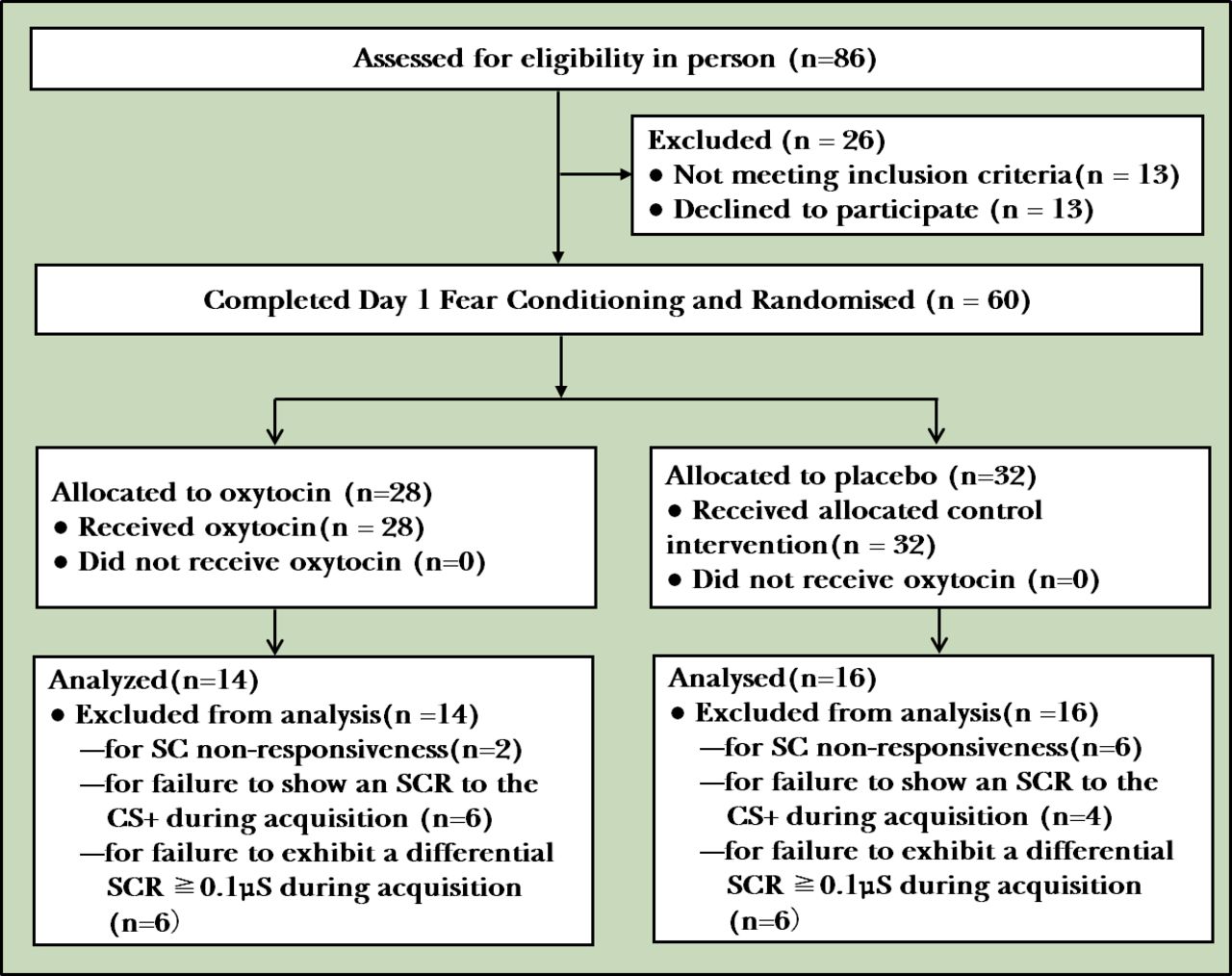
CONSORT flow diagram of participants. CONSORT, Consolidated Standards of Reporting Trials; SC, skin conductance; SCR, skin conductance response; CS, conditioned stimulus.
Sociodemographic, clinical and psychophysiological measures
The two groups did not differ significantly on any of the sociodemographic or clinical variables. To ensure our exclusion assumptions did not impact results, we replicated our analyses including all participants regardless of whether or not the respective SC criteria were met; this did not change the results (data not shown).
Baseline physiological levels, debriefing and shock levels
No significant differences in the levels of shock selected or the resting baseline SC level (measured by the SC preceding the first CS during habituation) were found between participants assigned to OT and those assigned to placebo (all p>0.05). Nine of the 14 OT participants and 14 of 16 placebo participants correctly identified the relationship between the CS+ and a shock (Fisher’s exact test, p=0.204, two-tailed).
Physiological response across all experimental phases
Day 1: habituation
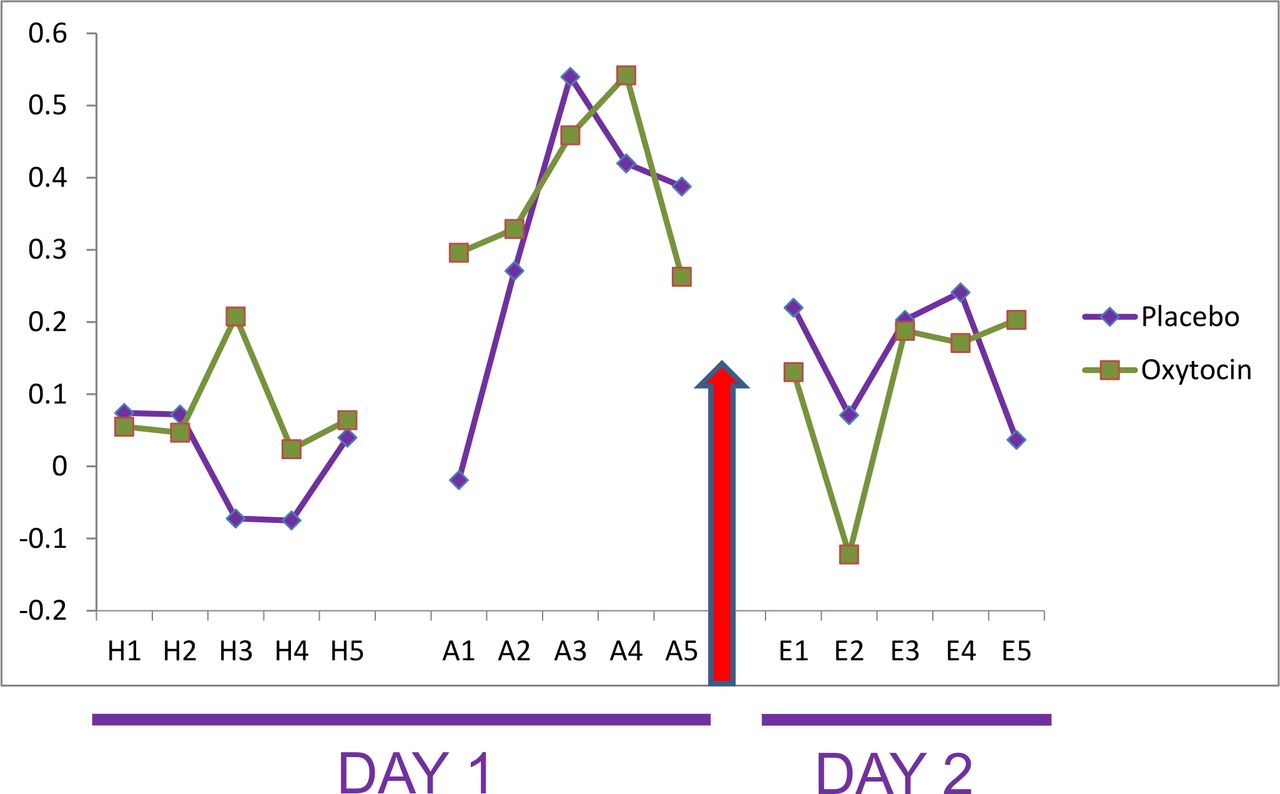
Each individual’s response to the first presentation of the CS+ and CS− was averaged to obtain the magnitude of the SC orienting response. Despite randomisation, prior to OT administration, individuals receiving OT exhibited a larger SC orienting response, compared with individuals receiving placebo (t=2.29, p=0.03). Further, an ANOVAR revealed that in the habituation phase the OT group exhibited a larger differential SCR (F(1, 28)=6.89, p=0.014) than the placebo group, which was mainly driven by differences in the third trial (t=2.96, p=0.013). However, the main effect of trials was not significant (F(4, 112)=0.74, p=0.57), nor was the interaction (F(4, 112)=1.57, p=0.187) (figure 2).
{kind=link}
{kind=link}
Differential scores (CS+ minus CS−) for the conditioned stimulus (CS) intervals for oxytocin versus placebo. Y-axis units are square-rooted μSiemens. A, acquisition trial; E, extinction trial; H, habituation trial.
Day 1: acquisition
During the ACQ, there was a trend towards increase in the differential SCR over time (F(4, 112)=2.1, p=0.085), followed by a slight decrease at the end of the phase, which is common for SCR in human conditioning studies. Further, supporting equivalence of the samples prior to OT administration, there was no significant main effect for treatment group (F(1, 28)=0.29, p=0.60) or for the group × trials interaction (F(4, 112)=0.70, p=0.59). Finally, there were no significant group differences in differential SCR at the end of conditioning (average of trials 4 and 5; t=−0.53, p=0.60).
Day 2: extinction
The first two extinction trials were used as a measure of fear acquisition. On day 2, there were no significant differences between the OT and the placebo groups for our primary measure of fear acquisition, the differential SCR averaged over the first two extinction trials (M=0.005, SD=0.394 vs M=0.146, SD=0.314, respectively; t=−1.092, p=0.28). Further, when examined across the first five extinction trials, the ANOVARs were not significant: trials (F(4, 112)=1.58, p=0.18), group (F(1, 112)=0.13, p=0.72), and group × trials (F(4, 112)=0.76, p=0.56). We also conducted a multiple regression to examine the association between group membership and differential SCR during the first two extinction trials while adjusting for the SC orienting response; this model was not significant (F(2, 27) =1.96, p=0.16).
As follow-up analyses, we examined SCRs to CS+ and CS− separately. For the first two extinction trials, the SCR to CS+ did not significantly differ between the groups; however, the SCR to the CS− was somewhat larger in the OT group compared with the placebo group (t=1.79, p=0.08). An ANOVAR of SCRs to CS+ trials revealed a significant effect of trials (F(4, 112)=7.04, p<0.001), but the group main effect (F(1, 112)=0.45, p=0.51) and group × trials interaction (F(4, 112)=0.41, p=0.79) were not significant. A similar pattern of results was found for the ANOVAR of SCRs to CS− trials.
Discussion
Main findings
To our knowledge, this study is the first to specifically examine the effect of OT on the consolidation of a de novo fear memory using a classical fear conditioning paradigm. Our analyses failed to show a statistically significant effect of 30 IU of double-blind intranasal OT versus placebo administered immediately after fear acquisition on fear expression as measured by the differential SCR to the first two extinction trials administered a day later. Further, there were no group differences for the SCR to the first two CS+ presentations during extinction or in the rate of extinction, as tested by the group × trials interaction effect.
Previous studies of OT’s effect on a conditioned fear response have used a 1-day procedure, in which acquisition and extinction phases were separated by less than 1 hour, and intranasal OT was administered sometime during this 1-hour window (eg, refs 17 19 37). These prior studies could not separately examine the effects of OT administration on fear consolidation versus extinction because the effect of intranasal administration of OT on circulating levels of OT has been reported to persist over 1 hour.38 By using a 2-day procedure in which acquisition and extinction were separated by approximately 24 hours, the present study was able to specifically examine the effects of OT on fear consolidation, as fear expression was tested at a time when the OT would have been completely metabolised.
Our study may have been underpowered as the effect size for the difference between OT and placebo on our primary outcome was modest (d=0.40) in the direction of OT. It is also possible that a higher dosage may have elicited a stronger effect on memory consolidation, as a dose-dependent relationship between administered OT dosing and plasma concentrations has been reported.38 It is also possible that the conditioned fear response was not sufficiently strong in our healthy sample so as to allow OT’s effect on memory consolidation to be detected, and a different finding might be present in those with pre-existing fear-related psychopathology.
While impairment in memory consolidation has been identified as a key component of PTSD pathophysiology (eg, refs 39 40), our findings, although preliminary, raise some doubt about the potential efficacy of intranasal OT administration on fear memory consolidation. OT has been proposed as a secondary prevention strategy for PTSD,41 with at least one large multisite randomised controlled trial currently ongoing.42 In this trial, trauma-exposed participants are randomised to receive either 40 IU of intranasal OT, or placebo, twice daily for 7.5 days, and assessed at 1.5, 3 and 6 months post-trauma for PTSD symptom severity. The study is testing the idea that OT may facilitate extinction learning in the first days after trauma exposure, as reported previously.19
The amygdala has been consistently found to be implicated in fear processing,43 and recent data suggest a modulating effect of OT on the amygdala. However, findings regarding the effect of OT on amygdala reactivity are equivocal. One recent study reported that intranasal OT administration dampens amygdala reactivity to presentations of emotional faces in individuals with PTSD,44 while a second study reported that OT administration in distressed, recently trauma-exposed individuals increases amygdala reactivity to emotional faces.45 Moreover, recent work found decreased functional connectivity between the amygdala and regulatory regions including the ventromedial and ventrolateral prefrontal cortices during script-driven imagery of the trauma, and increased self-reported PTSD symptom severity after trauma imagery.46 Although not statistically significant, in our sample individuals who received OT tended to exhibit a smaller differential SCR during extinction, compared with those who received placebo. However, this difference was not accounted for by a smaller SCR to CS+, which would have reflected weaker fear consolidation, but rather by a larger SCR to CS−. This could suggest a decreased ability to differentiate between CS+ and CS−, and thereby a decreased ability to identify the CS− as a ‘safety’ signal. This latter possibility is consistent with the reported effect of OT on reducing the functional connectivity between amygdala and ventromedial prefrontal cortex during script-driven imagery of non-traumatic scripts.46
Limitations
Our study was limited by a small sample size due to a large number of participants who were excluded from the analyses because they did not meet SC reactivity inclusion criteria. While sex has been suggested to moderate the effect of OT on fear processes,47 our relatively small sample precluded such an examination. Other limitations include group differences in SC orienting response and differential SCR during habituation prior to study drug administration, despite randomisation. Again, these unexpected differences are likely attributable to the small sample size. Prior studies have used a range of different dosages,48 and it is also possible that 30 IU might not have been an optimal dose. Finally, our study examined psychiatrically healthy individuals with normal memory functioning. It is possible that intranasal OT may be a more potent disruptor of fear memory consolidation in individuals with putatively dysfunctional memory, such as trauma-exposed individuals or those suffering from PTSD.
Implications
In conclusion, our results suggest that 30 IU intranasal OT administered in a double-blind fashion immediately after fear conditioning was not associated with decreased differential SCR at the beginning of extinction conducted 24 hours later.
References
Dr. Elizabeth Hoge is the director of the Anxiety Disorders Research Program and an associate professor of Psychiatry at Georgetown University Medical Center. Her research focuses on treatments for anxiety disorders, including medication, psychotherapy and mindfulness; improving resilience, and the examination of biomarkers of anxiety, stress, and trauma. She has over 45 publications in the psychiatric research literature and is on the Scientific Council of the Anxiety and Depression Association of America.

Dr. Eric Bui is an assistant professor of psychiatry at Harvard Medical School and the Center for Anxiety and Traumatic Stress Disorders and Complicated Grief Program at the Massachusetts General Hospital. He received his M.D. from the University Paul Sabatier, Toulouse, France, where he also completed a Ph.D. in Neurosciences. His research focuses specifically on understanding the mechanism and improving the treatment of anxiety and stress-related conditions. He currently serves on the Board of Directors of the International Society for Traumatic Stress Studies, and as Associate Editor of the European Journal of Psychotraumatology. To date, he has published over 120 scientific articles and book chapters in the field of anxiety disorders, posttraumatic stress disorder, complicated grief and related disorders.

Footnotes
EH and EB contributed equally.
Contributors EB and EH are co-first authors of this article. EH designed the study, oversaw all aspects of the research, drafted portions and substantively edited all drafts of the manuscript. EB conducted the analyses and produced the first draft of the manuscript. PR assisted in the literature review and data collection. SO contributed to data analyses and interpretation. RR interpreted the results and prepared the manuscript. RO contributed to data collection and interpretation of the results. NS contributed to study design, data interpretation and editing of the manuscript.
Funding Center for Integration of Medicine and Innovative Technology(ID: 001589).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The Massachusetts General Hospital Institutional Review Board approved all study procedures.
Provenance and peer review Commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.