Article Text

Abstract
The population of early-onset Alzheimer’s disease (EOAD) accounts for 1%–2% of the total population of Alzheimer’s disease, and genetic mutations are more common in EOAD. The first symptom of the patient in the present case report was the decline in memories of recent events, and the disease progressed rapidly in the following 2 years. Genetic testing has revealed the presence of genetic mutations (c.A479G, p.N160S) of ACE, which causes the 160th codon of the ACE protein to change from aspartic acid to serine, and at the same time genotype of apolipoprotein E (APOE) is ɛ3/ɛ4. We think that this patient carries the mutation type of the sensitive gene ACE and the risk gene APOE of Alzheimer’s disease, and this is the reason why the disease progressed rapidly. Moreover, we discussed ACE genetic mutation’s meaning in EOAD progression.
- mental health
- mental processes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
With the consistent increase of the ageing population, people’s awareness of Alzheimer’s disease (AD) has aslo increased, but our knowledge about its aetiological cause is still very limited. AD functional genomic studies indicate that age of onset, brain atrophy, cerebrovascular haemodynamics, brain biological electrical activities, cognitive function impairment, apoptosis, immune functions, lipid metabolism disorder and amyloid deposition are all related to AD genes.1 In a study of participants with AD, 1%–2% have early-onset Alzheimer’s disease (EOAD), and it is likely that genetic mutations are more common in EOAD.2 Research on families and twins has shown that mutations of the APP, PSEN1 and PSEN2 genes might lead to EOAD.3 With the emergence of genome-wide association studies and next-generation sequencing, more than 20 risk loci affecting AD have been identified. It is estimated that these loci explain about 28% of the hereditary risk, 30% of the family risk and more than 50% of the risk of recurrence in siblings with AD, and these are mainly because of the powerful influence of apolipoprotein E (APOE) ɛ4.4 The genotype of APOE is the most powerful hereditary risk factor, and ɛ4 allele is the risk factor for AD.5 On the other hand, according to a meta-analysis, PS1 1/2 genotype and ACE I/D genotype significantly increase the risk of suffering from AD in the Chinese population.6 If the basic mechanism and the potential neurodegenerative molecular mechanism of EOAD’s onset can be better understood, the development of effective diagnoses and treatment strategies will be promoted.
Experimental materials and methods
Genetic/biochemical procedure
Total genomic DNA was made from peripheral blood based on the standard method, and the quality of DNA was verified with Qubit V.3.0 (Thermo Fisher, USA) and gel electrophoresis method. The whole exon sequencing was verified with HiSeq X Ten (Illumina, USA), and we found a missense mutation on the ACE gene with second-generation sequencing and biological data analysis techniques when we focused on the analyses of AD-related genes. Following this, the verification with the first-generation Sanger sequencing was passed (upstream primer: 5’ –TCAGAAGTGATCCGGTCACA-3’; downstream primer: 5’ –GGAAGTCCACTTGCCAAGAG-3’; annealing temperatures were 60.2℃ and 59.8℃, respectively; the amplification product’s full length was 454 bp). In addition, we also tested the genotypes of APOE genes at two sites: rs429358 and rs7412. The mutation prediction software MutationTaster was employed to predict the pathogenicity of mutant genes (http://www.mutationtaster.org/).
Protein structure prediction
As the crystal structure of wild-type ACE protein is still unknown, we have found that sequences with a degree of identity over 30% can be used for homology modelling by searching for homologous sequences of ACE amino acid (1306aa) on the National Center for Biotechnology Information (NCBI) Basic Local Aligment Search Tool (BLAS7T) website in the Protein Data Bank (PDB) database. Therefore, we modelled the wild type and variant type of ACE protein with SWISS-MODEL.8 The predicted model was verified with multiple tools, including Verify 3D,9 ERRAT10 and PROCHECK.11
Results
Case introduction
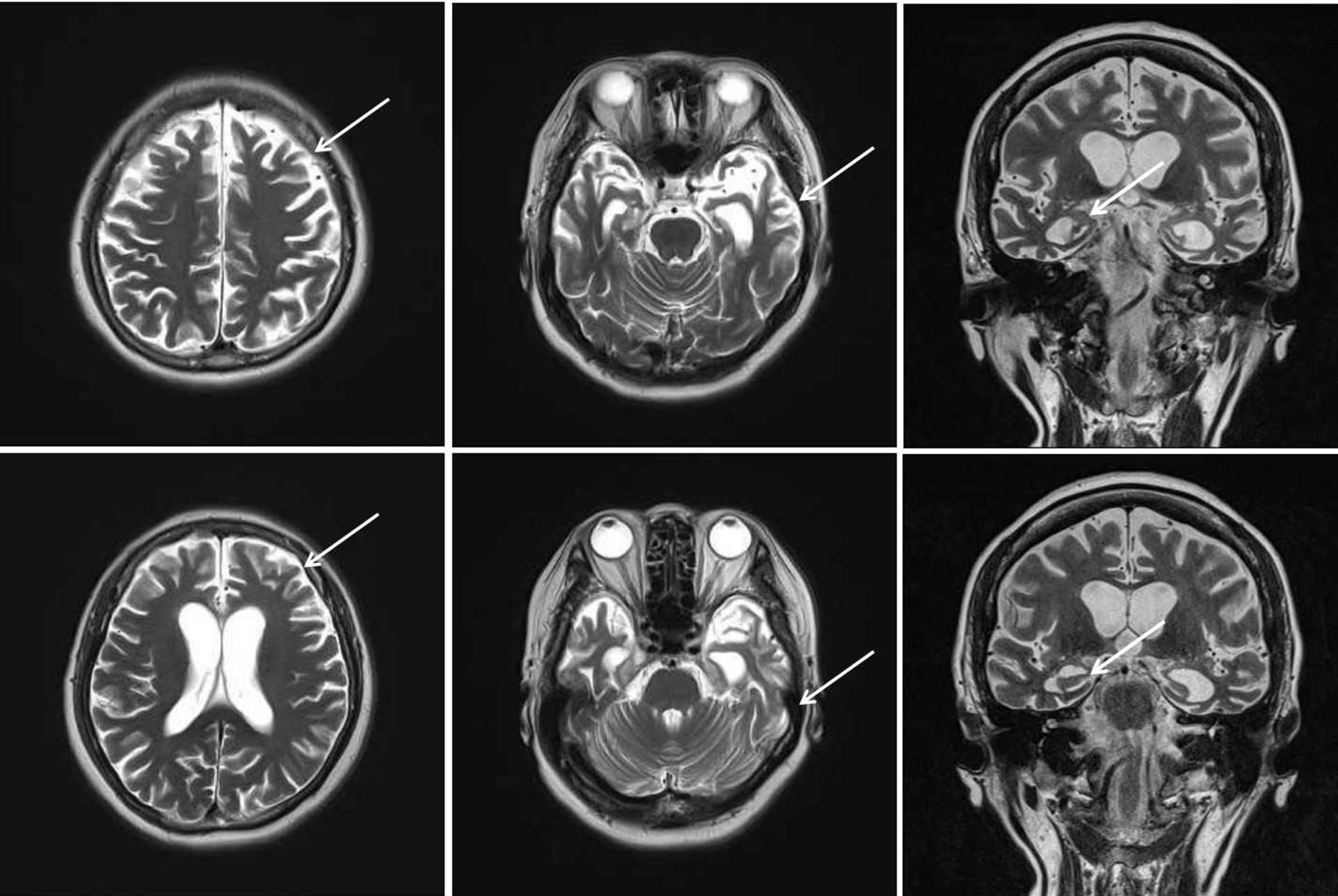
The case is a 63-year-old female Han Chinese patient with a college education. She worked in fashion design before she retired as a mental health worker. She has been living in the city for a long time. Two years ago, she started experiencing a decline in memory and feelings of agitation. One year ago, she started to feel dizziness and could not walk steadily. Her loss of memory was more obvious, and she would immediately forget what she said a minute ago. In addition, she also felt significant anxiety. After this, the disease progressed rapidly. She often got lost, and there was a serious decline in her work and life functioning. She has more than 10 years of history of hypertension, with the highest record of 180/100 mm Hg. Her blood pressure was normal when she retired, so she had not taken any medication to lower blood pressure. She did not have any other physical illness. Her mother has a history of dementia with an onset age of 78, and she is now 87 years old.,During the mental examination,she was in an awake state, her clothes were neat and tidy. She was passive when contacted, had flat affect, no disorganised behaviour, no delusions, hallucinations or perceptual comprehension disorder detected, no logical thinking or thought content disorders detected. She had a lack of thought content, difficulties in communicating, relatively dull emotional responses, and avolition. She performed poorly on assessments of calculation, common sense and comprehension. She had an overall lack of self control. Her neurological system examination showed no abnormal results. The patient was evaluated with related scales on 20 May 2016, and the results showed a score of 9 on the Mini-Mental State Examination (MMSE) (table 1A) and a score of 9 on the Montreal Cognitive Assessment (MoCA) (table 1B). Her score on the Hopkins Word Learning Test was 0, and her score on delayed memory was also 0. The Alzheimer’s Disease Assessment cognition subscale showed the following: word recall task: 10; object or finger naming: 7; following orders: 4; structural connection: 0; intentional connection: 0; orientation: 4; word recognition: 12; recall test instruction: 0; verbal language ability: 0; difficulty in finding words: 0; verbal language comprehension: 0; and attention: 0. Her score on the Activities of Daily Living Scale was 35. The degree of Global Deterioration Scale was 4. Her Hachinski Ischemic Score was 4. On 10 May 2018, the patient was evaluated with scales again, and the results showed a score of 0 on MMSE (table 1A) and a score of 0 on MoCA (table 1B). Laboratory examination included brain MRI, which showed brain atrophy (relatively significant in the frontal lobe) and fourth-degree atrophy in the coronal hippocampal area (picture taken on 20 May 2016) (figure 1) .
Scores on MMSE and MoCA scales from different evaluation dates
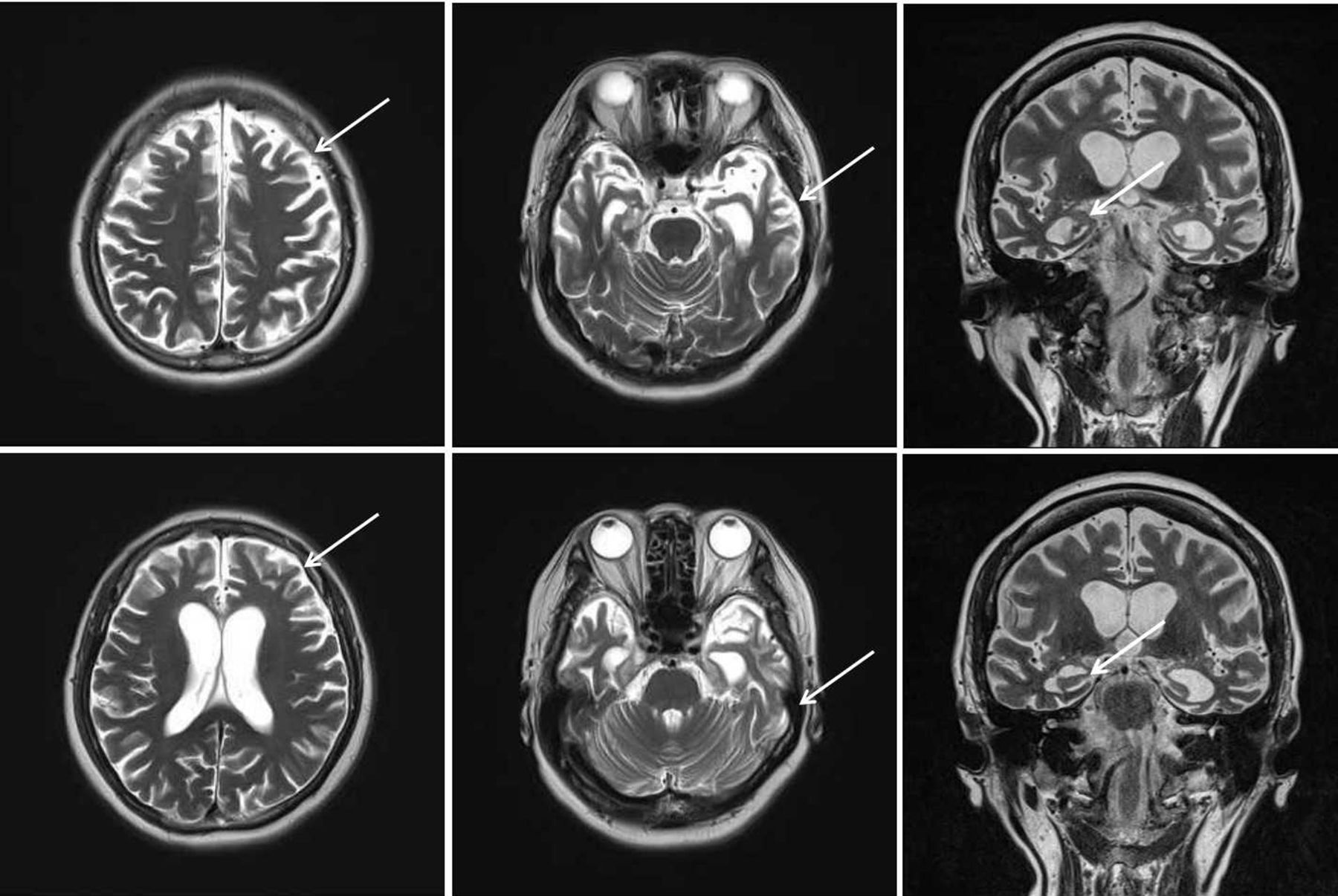
Brain MRI transverse section shows brain atrophy (relatively significant in the frontal lobe) and a fourth-degree atrophy in the coronal hippocampus area (picture taken on 20 May 2016).
Genetic analysis
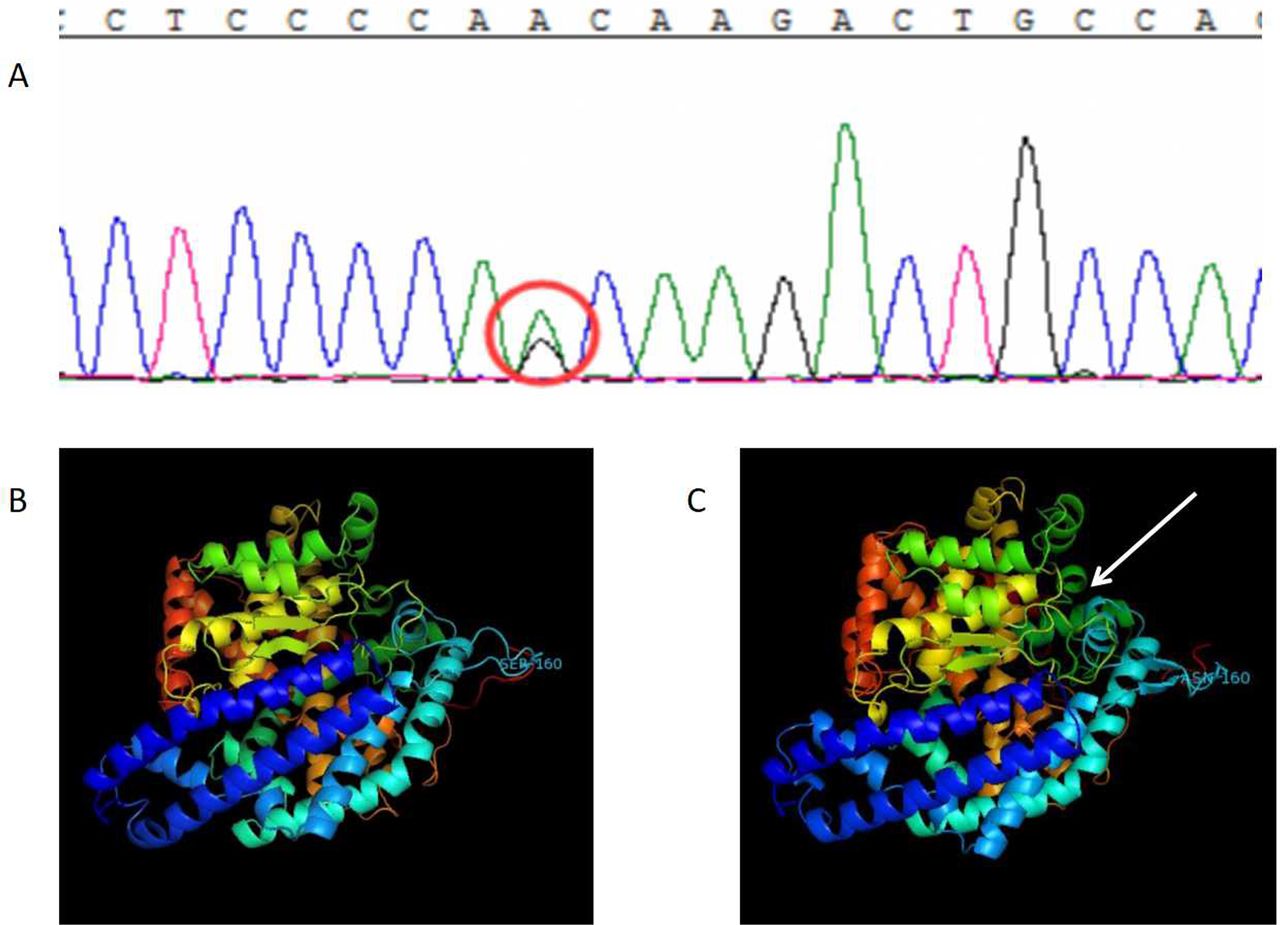
The subject’s degenerative-related exon was captured with second-generation sequencing, and a base change c.A479G of the subject’s third exon of ACE gene was detected. This base change caused the 160th codon of its encoding protein to change from aspartic to serine (figure 2A). This mutation was included in the Single Nucleotide Polymorphism Database(dpSNP) (rs117134739), but not in the 1000 Genomes database. Its frequency in the ExAC database is 0.001579. Bioinformatics software (PolyPhen-2, SIFT and MutationTaster) predicts that it might cause diseases.
{kind=link}
{kind=link}
DNA results and the ACE protein model. (A) There is a base change c.A479G on the third exon of ACE gene, and this base change has caused the 160th codon of its encoding protein to change from aspartic to serine. (B) The wild-type ACE protein structure and (C) the variant ACE protein structure. After the 160th amino acid was changed from aspartic to serine, the nearby areas changed from the original β-fold to the irregular curl, which caused an obvious change in partial protein structure.
Protein structure modelling
The variant ACE protein contains 1306 amino acids, of which the 160th amino acid was changed from aspartic to serine. With the SWISS-MODEL method, the template used for wild-type ACE was 5am8.1.A (figure 2B) and the sequence homology was 99.03%, while the template used for variant ACE was 4bzs.1.A (figure 2C) and the sequence homology was 99.68%. The model was verified by multiple examination tools. First of all, as for the wild-type ACE protein model, the scores on Verify 3D and ERRAT were 93.37% and 97.63%, respectively, and in PROCHECK 95.1% and 4.9% of amino acids were in the core and acceptable areas, respectively. As for the variant ACE protein model, the scores on Verify 3D and ERRAT were 96.85% and 97.27%, respectively, and in PROCHECK 93.8% and 5.6% of amino acids were in the core and acceptable areas, respectively. Verification methods mentioned above predicted a good quality of this model.
In the variant ACE protein model, it can be seen that after the 160th amino acid was changed from aspartic to serine, the nearby areas changed from the original β-fold to the irregular curl, which caused an obvious change in partial protein structure, thereby influencing protein function.
Discussion
AD is characterised by the gradual loss of memory, cognition and activities of daily life.12 The disease of the patient of interest progressed rapidly, and her overall cognitive functions were impaired within 2 years. The main symptoms were a severe decline in memory and an almost total loss of comprehension ability and executive ability within 2 years, acompanied by aphasia, loss of recognition,disuse. According to the results of the brain MRI, it was found that there was evidence of brain atrophy which was significant in the frontal cortex and a fourth-degree atrophy detected in the coronal hippocampus. Furthermore, this patient has a positive family history of dementia and a 40-year history of hypertension. The genetic testing showed that there was a base change c.A479G in the third exon of ACE gene, and this base change caused its encoding protein’s 160th codon to be changed from aspartic to serine. The genotype of APOE gene was ɛ3/ɛ4, so the symptoms of this patient were considered to be related to the genetic mutation.
The main pathological feature of AD is the formation of toxic amyloid due to abnormal processing of the amyloid peptides of the amyloid precursor protein (APP). It is a type of amyloid deposit that eventually leads to the disease caused by extensive destruction of neurons in the brain, which is similar to abnormal cholesterol deposition causing atherosclerosis. The renin-angiotensin system (RAS) affects the blood vessels, metabolic homeostasis, amyloid metabolism, and learning and memory. As a key RAS protein, ACE contains many variant mega genes and codes associated with AD13 because ACE can degrade the β amyloid-like protein, which is a pathological symbol of AD, thereby inhibiting its aggregation. The level and active portion of ACE may be regulated by the insertion and deletion polymorphism of ACE gene.14 Recent evidence has indicated that AD has different vascular risk factors from other prevalent dementia types, and it may cause the deterioration of cerebrovascular function, thereby increasing neurodegeneration and premature neuronal deaths due to reduced cerebral perfusion.15 In the mouse model of AD, ACE 10/10 phenotype provides significant protection for AD pathology, including reducing inflammation, reducing the burden of neurotoxic amyloid-βprotein and maintaining cognitive functions. These studies have shown that in order to better protect against many different types of pathological damage, including cognitive decline observed in AD animal models, the increased monotype ACE expression in mice alters immune responses.16 ACE is widely expressed in the brain, so it may play a role in the pathogenesis of dementia.17
APOE mainly affects the pathogenesis of AD by affecting amyloid-like β protein, and the aggregation of substantial plaques and the β protein in cerebral amyloid angiopathy triggers neuroinflammation.18 In addition, APOE may affect neuroinflammation, cerebrovascular integrity, metabolism, synaptic plasticity and transcription directly with the independent mechanism of amyloid-like β protein.
The patient of interest in this article has a base change c.A479G on the third exon of the ACE gene, and this base change has caused the 160th codon of its encoding protein to change from aspartic to serine. This phenomenon may lead to the dysfunction in the RAS, which caused the patient to have high blood pressure for nearly 40 years. In the mean time, alterations in the ACE gene may lead to an increase in toxic brain amyloid deposition. On the other hand, because the aggregation of amyloid-like β protein in human brains follows the pattern of APOE4 > APOE3 > APOE2,19 the ɛ3/ɛ4 type of APOE gene in the patient promotes the aggregation of amyloid-like β protein, thereby further aggravating the neuroinflammation, leading to a significant decline in cognitive functions and the rapid progression of the disease.
This case cannot have regular follow-ups because the patient left the country soon after. Furthermore, it is hard to obtain blood samples from her family members to conduct genetic analysis, so we cannot explore whether the ACE gene and APOE gene exist in her family members or not, and it is also hard to track whether her family members suffer from EOAD or not.
References
Jing Ni obtained her bachelor's degree from Southeast University School of Medicine in 2003. She is currently working as an attending doctor at Shanghai Huangpu District Mental Health Center Psychiatric Department. Her research interest includes elderly psychiatry.

Footnotes
Contributors JN was responsible for writing the paper. LS was responsible for data collection and analysis. XL was responsible for quality control. SX was responsible for providing guidance on study design.
Funding Shanghai Huangpu District research project 'Correlation study of different dimensions of mental behavior symptoms in patients with dementia and the mood disorder in their caregivers' (project approval number: HKM201721).
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.