Article Text

Abstract
Background Evidence from clinical and preclinical studies has demonstrated that stress can cause depressive-like symptoms including anhedonia and psychomotor retardation, namely, the manifestation of motivational deficits in depression. The proximate mediator of linking social-environmental stress with internal motivational deficits remains elusive, although substantial studies proposed neural endocrine mechanisms. As an endogenous danger-associated molecule, high mobility group box-1 (HMGB1) is necessary and sufficient for stress-induced sensitization of innate immune cells and subsequent (neuro)inflammation.
Aim This review aims to provide evidence to unveil the potential mechanism of the relationship between motivational deficits and stress in depression.
Methods We reviewed original case-control studies investigating the association between HMGB1-mediated inflammation and stress-induced depression. The literature search of Pubmed and Web of Science electronic database from inception up to March 28th, 2019 were conducted by two independent authors. We performed a qualitative systematic review approach to explore the correlation between HMGB1-mediated inflammation and anhedonia/psychomotor retardation in depression.
Results A total of 69 studies based on search strategy were retrieved and seven eligible studies met the inclusion criteria. Studies showed that HMGB1 was implicated with depressive-like behaviors, which are similar with motivational deficits. Furthermore, HMGB1-mediated inflammation in depressive-like behaviors may be involved in Nod-like receptor family pyrin domain containing three (NLRP3) inflammasome and proinflammatory cytokines, abnormal kynurenine pathway and imbalance between neuroprotective and neurotoxic factors.
Conclusions We found that stress-induced inflammation mediated by HMGB1 may affect motivational deficits through regulating dopamine pathway in corticostriatal neurocircuitry. The systematic review may shed light on the novel neurobiological underpinning for treatment of motivation deficits in depression.
- neuroinflammation
- motivational deficits
- HMGB1
- corticostriatal circuitry
- depression
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Major depressive disorder (MDD) is clinically characterised by motivational deficits.1 Motivational deficits are manifested in the symptoms of anhedonia and psychomotor retardation. Anhedonia refers to the reduced motivation or ability to experience pleasure, and psychomotor retardation is known as a slowing-down of thought and a reduction of physical movements.2 Evidence from clinical and preclinical studies has demonstrated that stress can trigger anhedonia and psychomotor deficits in vulnerable individuals.3 4 Chronic and acute stressors are among the strongest proximal risk factors for MDD.5 Consistent with this notion, chronic unpredictable mild stress (CUMS) and social defeat stress have been used to establish animal models of depression, resulting in anhedonia-like behaviours and increasing immobility time.6–8 However, few studies directly investigate the impact of stress on brain reward processing and explore the potential mechanisms in stress-induced motivational deficits. Some studies revealed that stress-induced motivational deficits may involve in the abnormal mesolimbic dopaminergic circuit,9 10 although the cause of such dysfunction remains elusive.
During the past decades, substantial evidence has demonstrated that exposure to stress can prime microglia and strengthen the neuroinflammatory response to the central nervous system (CNS) and peripheral inflammatory challenge.11 Accordingly, CNS and peripheral inflammation has been assumed to be associated with the psychopathology of MDD.12 Therefore, we review clinical and preclinical evidence to clarify the relationship between stress-induced motivational deficits in MDD and the potential signal pathway in the stress-inflammation-mesolimbic reward circuit.
The association between stress and neuroinflammation: the role of the danger-associated molecular pattern HMGB1
Psychological and physical stress can potentiate the proinflammatory response of microglia to subsequent immune stimulators through releasing endogenous danger molecules known as danger-associated molecular patterns (DAMPs).11 These DAMPs are recognised by pattern recognition receptors (PRRs) in the cellular surface, such as toll-like receptors (TLRs) and the receptor for advanced glycation end products (RAGE). CNS and peripheral innate immune system become activated in response to the recognition of DAMPs-PRRs signalling.13 However, the proximate DAMPs mediators by which stress primes microglia still need to be elucidated. One previous review showed that several DAMPs, including S100 proteins, high mobility group box-1 (HMGB1), heat shock proteins, ATP and uric acid can result in depressive-like behaviours in stress-induced depression models.14 In particular, HMGB1 is considered as master regulator of innate immunity.15 Previous studies suggested that HMGB1 signalling is necessary and sufficient for stress-induced sensitisation of microglia, then amplifying the expression of the NLR family pyrin domain containing 3 (NLRP3) inflammasome and proinflammatory cytokines.16 Moreover, HMGB1 has been involving in several neuroinflammatory conditions, such as Alzheimer’s disease17 and brain ischaemia injury.18 Therefore, the scope of our review focuses on the HMGB1 as a potential mediator of stress-induced neuroinflammation in MDD.
The association between neuroinflammation and motivational deficits mediated by brain structure and function
Experimental cytokine (eg, injection of typhoid vaccination or LPS) could quickly trigger fatigue, depressed mood, lack of interest and psychomotor retardation in healthy individuals, suggesting the underlying alterations of neural activity in specific regions. Previous studies suggest that stress is implicated with the aetiology of MDD through activating neuroinflammation processes and then involving structural and functional alterations of specific brain regions.19 Neuroinflammation exerts direct detrimental effects on brain structure and function by mediating neuronal damage/degeneration, glial activation, mitochondrial dysfunction, demyelination and endothelial damage.20 Moreover, these inflammation-induced behavioural changes have been assumed to be associated with the alterations of neurotransmitters metabolism (serotonin, dopamine (DA) and norepinephrine)21 22 and the activation of indoleamine 2,3-dioxygenase (IDO) in kynurenine pathway (KP),23 which are consistent with the psychopathology of MDD. Neuroimaging techniques have been advanced to confirm the link between inflammation and depression mediated by the alterations in neural regions associated with the regulations of motivation and reward, including prefrontal regions and ventral striatum,24 25 and the regulation of emotion, including amygdala, anterior cingulate cortex and prefrontal cortex.26 27
As mentioned above, evidence has showed that stress can cause depressive-like symptoms including anhedonia and psychomotor retardation, which are common in patients with MDD.28 Similar findings were observed in depressive-like animal models by reductions in saccharin preference and increased immobility time.29 However, little is known about the underlying mechanism of the relationship between motivational deficits and stress in depression. One candidate neurobiological mechanism, overactive inflammation, was supposed to be associated with motivational deficits in MDD.30 Therefore, we hypothesised that stress-induced neuroinflammation mediated by HMGB1 may be implicated with the alterations of corticostriatal reward neurocircuitry contributing to anhedonia and psychomotor retardation in MDD.
Methods
Inclusion criteria
We recruited original studies investigating the association between HMGB1-mediated inflammation and stress-induced depression. Depressive-like behaviour in an animal model was induced by stress, such as CUMS. The studies recruited into this review were case-control studies that compared the expression levels of HMGB1 in stress exposure group and control group. Only studies written in English were recruited into our review.
Exclusion criteria
Literature reviews, systematic reviews, meta-analysis and conference or meeting abstracts were excluded. Articles where a depressive-like animal model was not induced by stress were also excluded.
Search strategy
The PubMed and Web of Science electronic databases were searched from inception up to 28 March 2019. The following search string was used: (depression OR major depressive disorder OR unipolar depression OR major depression) AND (HMBG1 OR high-mobility group box 1 protein). Meanwhile, we manually searched the reference lists of each eligible article to identify additional studies.
Data extraction
For each eligible article, we extracted and recorded the following important information: first author and year of publication, animal models, the changes of expression levels of inflammatory cytokines, depressive-like behaviours and antidepressants-like target.
Data synthesis
The high level of heterogeneity of HMGB1-mediated inflammatory biomarkers was observed in the present review. Thus, quantitative analysis (eg, meta-analysis) was not conducted. Instead, we performed a qualitative systematic review approach to explore the correlation between HMGB1-mediated inflammation and anhedonia/psychomotor retardation in depression.
Results
Characteristic of included studies
Our search found 69 studies based on the search strategy and manually checking the reference lists of searched articles. After title/abstract screening and full text reviewing, only seven eligible studies met the inclusion criteria. The flowchart for search and study selection is illustrated in figure 1. The characteristics of these seven studies are listed in table 1.
Systematic-summarised studies of HMGB1-mediated inflammation in depressive-like animal models

The flowchart of search and study selection.
Association between HMBG1-mediated inflammation and depression
All published original research involving HMGB1 in stress-induced depressive animals are listed in table 1. Studies have showed that HMGB1 is implicated with depressive-like behaviours (eg, reduced saccharin preference and reduced locomotor activity), which are similar with motivational deficits (eg, anhedonia and psychomotor retardation) in human studies. HMGB1 elicited anhedonic behaviour via inducing TNF-α and then promoting neuroinflammatory response activation,31 32 whereas glycyrrhizic acid and ethyl pyruvate, inhibitors of HMGB1, could improve depressive-like behaviours.8 31 Moreover, Weber et al16 established inescapable tail shocks animal depressive models to explore whether HMGB1 is a potential mediator of stress-induced microglia priming.16 These findings showed that HMGB1 was sufficient to prime microglia and subsequent NLRP3 inflammasome and proinflammatory cytokines. Moreover, HMGB1 mediated depressive behaviour through restraining the activated enzymes in KP, such as IDO.8 19 Noticeably, IDO, the rate-limiting enzyme of KP, is highly inducible by proinflammatory cytokines (eg, TNF-α, interleukin 1β). Cheng et al32 also found that stress-induced depression-like behaviour is mediated by GSK3-dependent TLR4 signalling, which further upregulated HMGB1 and activated nuclear factor κB (NF-κB) and NLRP3 inflammasome.32 Moreover, persistent microglial HMGB1-RAGE expression increase susceptibility to chronic stress-induced priming of depressive-like behaviours.13 Therefore, HMGB1 might be a plausible danger signalling mediator of stress-induced depressive-like behaviours, which is involved in DAMPs pattern recognition, including HMGB1-TLR4 signalling33 and HMGB1-RAGE signalling.34
Discussion
In this systematic review, we aimed to explore the association between HMGB1-mediated inflammation and motivation deficits in depression. Numerous preclinical studies strengthen our understanding of the HMGB1-mediated inflammation in depression. Although there is a lack of evidence in clinical studies of HMGB1, evidence from aforementioned reviewed studies indicated that, HMGB1, although not mutually exclusive interpretation, may play a critical role in the regulation of stress-induced neuroinflammation14 and be further involved in the motivation dysfunction through altered corticostriatal circuitry.21
The association between stress and MDD involving neuroinflammation
Substantial clinical studies have suggested that stress is associated with upregulated inflammatory activity.35 36 Consistent with this notion, laboratory-based preclinical studies clarified that acute and chronic stress induced the higher levels of interleukin-6 (IL-6), and C reactive protein in serum and brain tissues, which in turn elicit profound changes of behaviour.37 Postmortem studies in MDD found that proinflammatory and anti-inflammatory cytokine gene expression were upregulated in the prefrontal cortex.38 Neuroimaging studies showed that increased inflammatory response was associated with abnormal corticostriatal reward circuitry in depression, which in turn correlated with increased psychomotor slowing and anhedonia.39 Therefore, there is no debate that motivational deficits have been implicated with neuroinflammation in MDD.
The role of the danger-associated molecular pattern HMGB1 in stress-induced motivational deficits
HMGB1 is a chromatin intracellular protein expressed in all tissues of mammals, which mostly locate in nuclei.40 In the peripheral, HMGB1 can be secreted from cells, macrophage and monocytes. In the brain, abundant HMGB1 can be expressed in neurons, microglia and astrocytes.41 HMGB1 can be passively released from necrotic cells. In addition, HMGB1 is also involved in active secretion from innate immunes cells in response to stress.31 It has emerged as one of the main mediators in the pathophysiology of autoimmune diseases and neuroinflammation,42 such as systemic lupus erythematosus, cerebral ischaemia, traumatic brain injury and seizure. The extracellular HMGB1 is an endogenous danger and priming signal that uses cellular-ligand PRRs, including TLR2, TLR4 and RAGE.14 Activation of PRRs can initiate cell proliferation and migration as well as proinflammatory cytokine synthesis through mitogen-activated protein kinases-NF-κB signalling pathway.43 Moreover, stress-induced neuroinflammation contribute to vulnerability to depression that involves upregulation of HMGB1, activated NF-κB and subsequent exaggerated NLRP3 inflammasome.32 44 The mechanism of HMGB1 under oxidative stress in depression was supported by the finding that redox forms of HMGB1, but not nonoxid-HMGB1, induced depressive-like behaviour mainly through neuroinflammatory response activation.45 As mentioned in table 1, HMGB1-mediated inflammation could induce depressive-like behaviours in animal models, such as decreased sucrose preference and increased immobility time in the tail suspension test and forced swimming test. Of note, these behaviours are similar to anhedonia and psychomotor retardations in depressed subjects, namely, the manifestation of motivational deficits.
Neurobiological basis of inflammation-induced motivational deficits in MDD: dopamine and corticostriatal neurocircuitry
Symptoms related to motivational deficits are difficult to treat by selective serotonin reuptake inhibitors and serotonin and norepinephrine reuptake inhibitors,46 indicating that other neurotransmitters may be implicated with inflammation-induced motivational deficits in MDD.47 Experimental studies in rats found that lesions of dopaminergic neurons in the mesolimbic system could cause anhedonic behaviours.48 In humans, similar findings have been observed that blocking DA reuptake or/and increasing DA release can improve motivational activity.49 50 Hence, substantial evidence demonstrated that DA dysfunction may contribute to the regulation of motivational deficits in depression.21 Consistent with this, neuroimaging and pharmacological studies found that dopaminergic transmission could enhance striatal activation and corticostriatal functional connectivity in depressive subjects.51
Given that dopaminergic neurons of the mesolimbic system involved in motivation and reward-behaviours, numerous neuroimaging studies have found a significant relationship between motivational deficits and the mesolimbic reward circuit, including prefrontal regions, ventral striatum, afferent and efferent projections (see table 2). Functional MRI studies revealed that decreased ventral striatum response and disturbance of orbitofrontal cortex were associated with anhedonia and fatigue mediated by inflammatory cytokines.39 52 Moreover, prefrontal cortex overactivity suppressed reward-motivated behaviours by modulating striatal activity.53 Positron emission tomography studies using the D2/3 receptor-selective radiotracer [11C] raclopride found that depressed subjects showed increased D2/3 receptor availability in the ventral striatum.54 As such, neuroimaging studies have provided some robust evidence that corticostriatal reward circuit has been involved in motivational deficits in depression.25
Summary of findings from neuroimaging studies exploring the effect of inflammation on glutamate/dopamine and/or reward circuitry
Candidate mechanisms of stress-induced neuroinflammation effects on motivational deficits through dopaminergic corticostriatal neurocircuitry
Exposure to stress can eventually result in a continuum of clinical manifestations, including anhedonia and psychomotor retardation. However, little is known about the potential mechanisms of the relationship between stress-induced neuroinflammation effects on motivational deficits in depression. Based on aforementioned studies, the relatively reliable notion can be drawn that HMGB1-mediated neuroinflammation may be involved in motivational deficits in depression. Several candidate mechanisms have been proposed, including regulating the synthesis, transportation and availability of DA, KP pathway modulating the expression level of glutamate, abnormal synaptic plasticity and neuronal dysfunction.
More specifically, growing evidence has demonstrated that HMGB1-mediated neuroinflammation exerts on the synthesis, transportation and availability of DA. After exposure to stress, microglia secreted reactive oxygen species (ROS) and nitrogen species (NOS), which may rapidly decrease neopterin and tetrahydrobiopterin (BH4) availability, resulting in inactivation of phenylalanine hydroxylase (PAH) and tyrosine hydroxylase (TH).55 As rate-limiting enzymes for DA synthesis, PAH and TH can catalyse phenylalanine into tyrosine and then gradually convert to DA. As such, DA synthesis is hampered by HMGB1-mediated inflammation via increasing oxidative stress. Moreover, further evidence reveals that HMGB1-mediated inflammation also affects the transportation and availability of DA. Given that vesicular monoamine transporter-2 (VMAT-2) exerts on the transportation of synaptic DA, biomarkers of inflammation may affect the release of synaptic DA by regulating the expression and function of VMAT-2. Consistent with this notion, proinflammatory cytokines can decrease expression of VMAT-2 in experimental studies.56 Hence, biomarkers of inflammation may also decrease DA signalling by reducing D2 receptor and increasing DA transporter (DAT).21 Consequently, HMGB1-mediated inflammation may interrupt the metabolism of DA, which can be supposed to impair the function of corticostriatal neurocircuitry and to result in motivational deficits.
Another mediator about regulation of DA function and corticostriatal neurocircuitry is glutamate. Magnetic resonance spectroscopy studies suggested that inflammatory cytokines may cause the increased concentrations of glutamate in the basal ganglia, which in turn associated with psychomotor slowing and anhedonia in patients with MDD.57 Furthermore, glutamate receptor antagonist ketamine could improve depressive symptoms in human and laboratory animal studies. One pathway by which glutamate neurotransmission involves in HMGB1-mediated motivational deficits may be indirectly mediated by KP rate-limiting enzymes and neurotoxic metabolites. Consistent with this, IDO inhibitor 1-mehtyltryptophan could attenuate anhedonia-like behaviour in an animal model.58 In human studies, an index of IDO activity (kynurenine/tryptophan) was associated with the severity of anhedonia in depressed subjects.59 Proinflammatory cytokines (eg, interferon-α, TNF and IL-6) upregulate the expression of vital enzymes (eg, IDO, kynurenine-3-monooxygenase) involved in the neurotoxic arm of the KP. As such, under the catalysis of immune-mediated activation of IDO and kynurenine-3-monooxygenase, tryptophan is converted to neurotoxic metabolites 3-hydroxykynurenine, 3-hydroxyanthranilic acid and quinolinic acid (QUIN).60 Particularly, increasing neurotoxic metabolite QUIN can heighten the activity of N-methyl-d-aspartate glutamatergic receptor and alter the glutamatergic neurotransmission, which subsequently causes glutamate excitotoxicity in the brain. Alternatively, another pathway is that HMGB1-mediated inflammation directly results in the increasing extracellular glutamate buildup.61 As mentioned above, microglia secreted ROS and NOS resulting in the increasing oxidative stress in the brain.55 The imbalance between neuroprotective factors and neurotoxic factors causes the dysfunction of glutamate exchange in cell membranes, which further increases the concentration of extracellular glutamate and the synthesis of DA. Thus, excessive glutamate neurotransmission and excitotoxicity may be involved in the pathophysiological progression of HMGB1-mediated motivational deficits in MDD.62
Moreover, another plausible mechanism of HMGB1-mediated motivational deficits involves abnormal synaptic plasticity and neuronal dysfunction. Therefore, dysfunction of DA in corticostriatal neurocircuitry may cause abnormal corticostriatal synaptic connections. Evidence from animal studies showed that chronic stress caused front striatal reorganisation including neuronal densities, number of intersections and dendrites morphology, which caused a bias in behaviours.63 Furthermore, LPS-induced inflammation-related depression was implicated in the alteration of brain-derived neurotrophic factor (BDNF) and its receptor, tropomycin receptor kinase B (TrkB) in frontostriatal regions.64 As a neurotrophic protein, BDNF plays a vital role in maintaining the survival of existing neurons and enhancing the growth and differentiation of new neurons and synapses. As mentioned above, HMGB1-mediated inflammation produces neurotoxic metabolites (eg, ROS, proinflammatory factors). Hence, excessive neurotoxic metabolites and abnormal BDNF-TrkB signalling may cause neuronal degeneration and alteration of synaptic plasticity in frontostriatal neurocircuitry, which in turn affect reward-related behaviours.
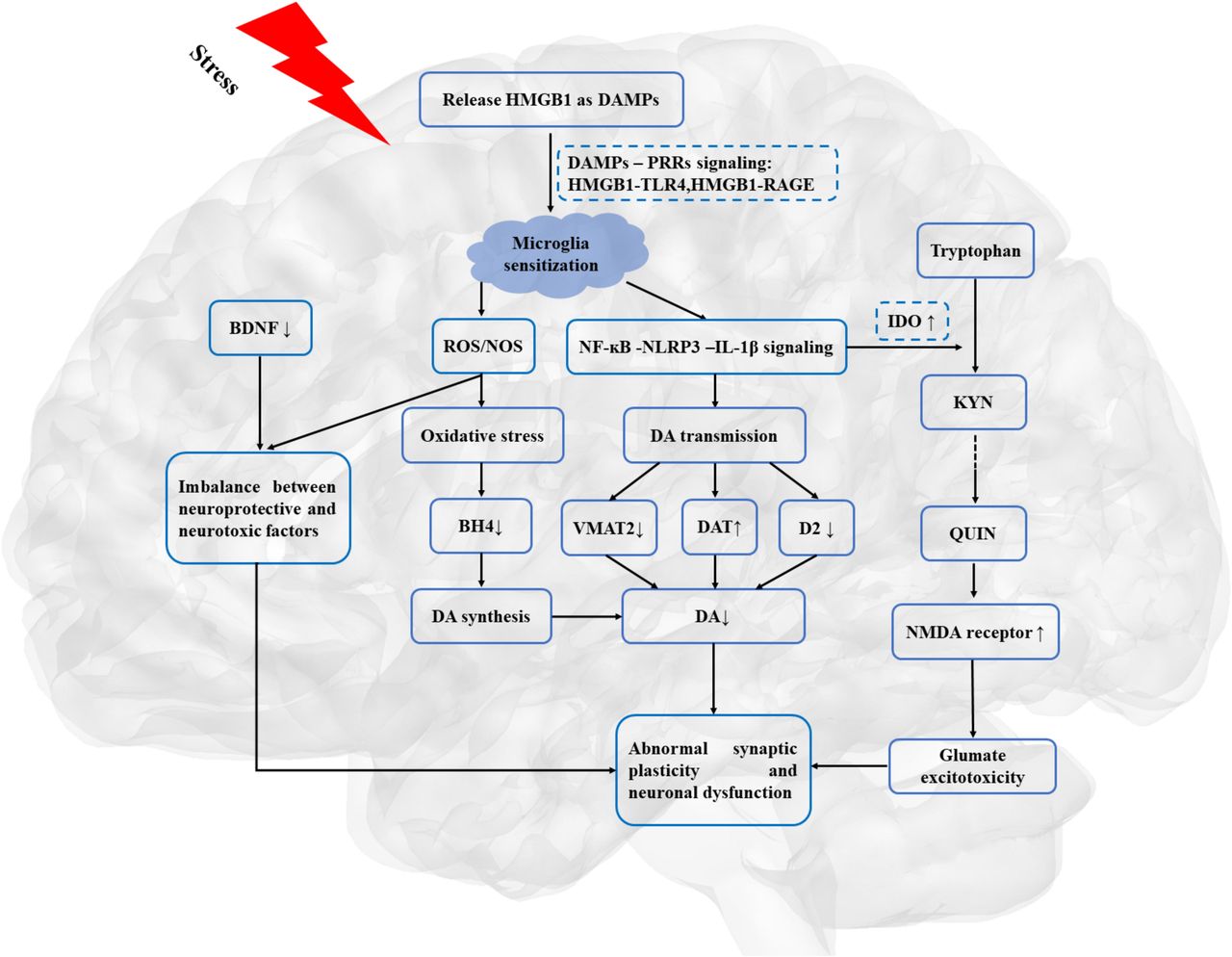
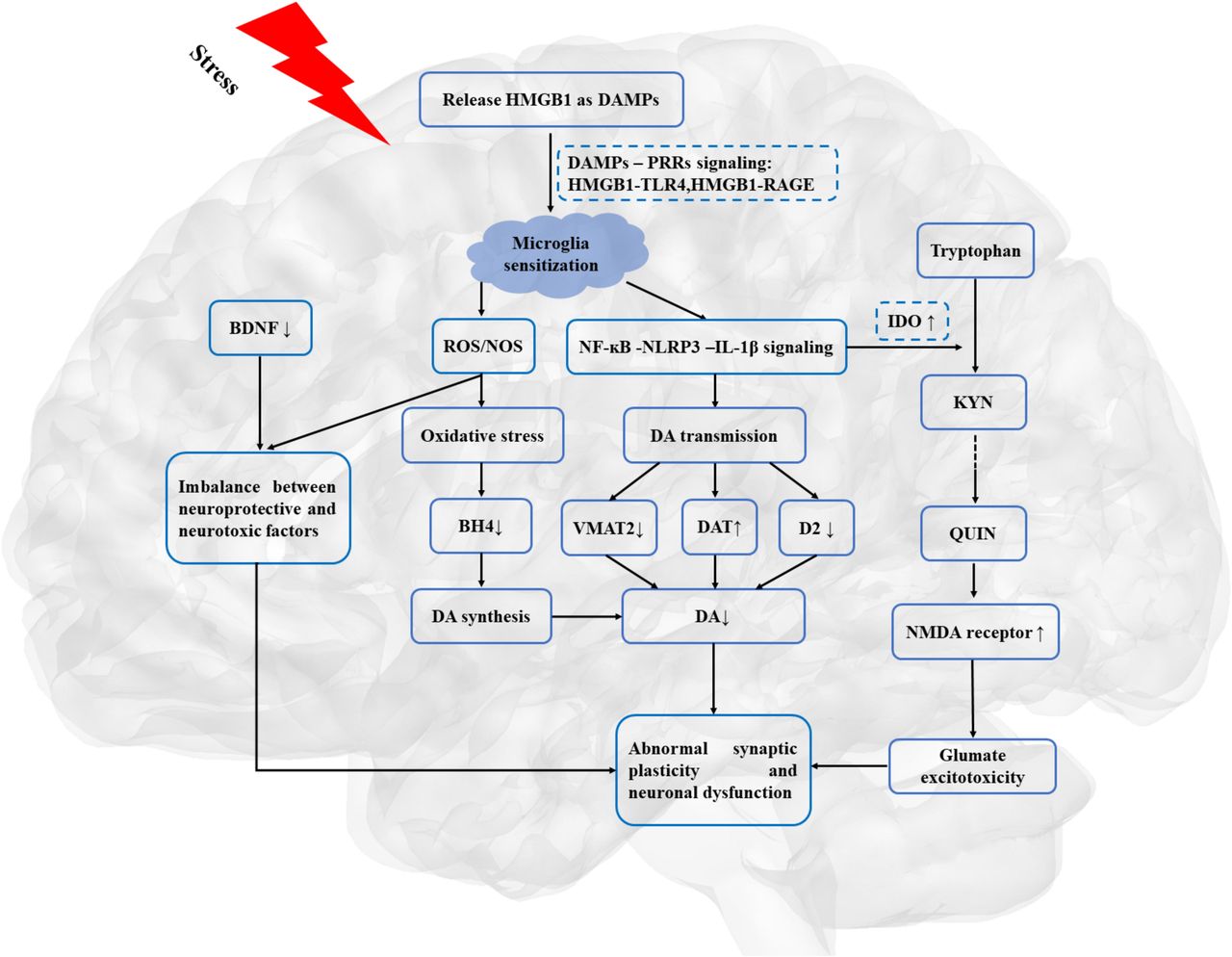
As illustrated in figure 2, HMGB1-mediated inflammation may lead to the dysfunction of DA neurotransmission and dopaminergic corticostriatal circuitry, which may provide the neurobiological basis for reduction in motivation in depression.
{kind=link}
{kind=link}
HMGB1, as a priming signalling, involved in the stress-neuroinflammation-mesolimbic dopaminergic pathway. BDNF, brain-derived neurotrophic factor; DA, dopamine; DAMPs, danger-associated molecular patterns; DAT, dopamine transporters; D2, dopamine receptor 2; HMGB1, high mobility group box 1; IL-1β, interleukin 1β; IDO, indoleamine 2,3 dioxygenase; KYN, kynurenine; NMDA, N-methyl-d-aspartate; NF-κB, nuclear factor κB; NOS, nitrogen species; PRRs, pattern recognition receptors; QUIN, quinolinic acid; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; TLR4, toll-like receptor 4; VMAT-2, vesicular monoamine transporter-2.
Discussion
This systematic review indicated that HMGB1 may shed light on the stress-neuroinflammation-mesolimbic dopaminergic pathway in the understanding of the pathopsychological mechanism underlying depressive behaviours, such as anhedonia and psychomotor retardation. The importance of HMGB1-mediated inflammation in depression may need to be stated explicitly, with a discussion of the potential implications for the understanding of the pathopsychology of MDD.
References
Huifeng Zhang graduated with a bachelor’s degree from Xinhua Hospital affiliated to Shanghai Jiao Tong University, school of medicine in 2017. She is currently studying for a master’s degree at the Shanghai Mental Health Center, Shanghai Jiao Tong University school of medicine, Shanghai, China. Her research interest includes major depressive disorder and bipolar disorder.

Footnotes
HZ and LD are joint first authors.
Contributors DHP, TS, and HFZ designed the protocol and searched the literature. HFZ and LD screened the studies and extracted the data. TS double-checked the extracted studies. HFZ wrote the first draft of the manuscript. DHP and TS have revised the main manuscript. All authors have approved the final version of the main manuscript for submission.
Funding This research was supported by the National Key Research and Development Program (Grant No. 2016YFC0906400) and Shanghai Science and Technology Committee (Grant No. 18JC1420304).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All the relevant data has been presented in Tables and Figures. No additional data are available.